


How It's Done: Genetic Technologies
- Aarushi Gupta

- Dec 22, 2019
- 8 min read
Updated: May 23, 2020
There are plenty of things to talk about when it comes to bioengineering DNA to produce desired effects. In the third and final part of my study, we'll discuss manipulating and cloning DNA and methods of DNA sequencing.
In this post, we'll discuss 4 processes that help. There are certainly more than that but my previous post got very long because I tried to fit as much information in it as possible. Before beginning, recombinant DNA means a DNA strand created using DNA pieces from 2 or more different sources.
1. Restriction Enzyme
2. Plasmids
3. Restriction Maps
4. Transformation
A restriction enzyme's function is to cut DNA at very specific sites. It does so by breaking the phosphodiester bonds between 3' and the phosphate of the next nucleotide. The need for this is high in bacteria. Bacteria don't have a complex immune system, so when they are attacked by a bacteriophage, the restriction enzymes act as an immune system and cut the virus' DNA into small pieces that can't go back together. This protects the cell from being affected by the DNA and from the possibility of lysis or bursting when the new viruses were ready to leave the cell.
Each restriction enzyme has a specific site of 4-8 nucleotides where it cuts, called the recognition site.
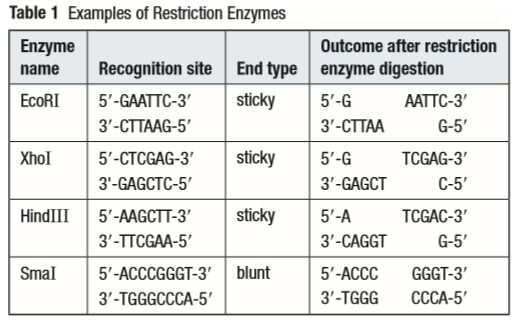
At the recognition site, depending on the enzyme, a sticky end can be made or a blunt end can be made. A sticky end is more favourable in genetic engineering. In a sticky cut, some nucleotides remain without a complementary strand. This allows scientists to use the same enzyme, cut a part of the DNA they need cut, and attach it to the bacterial DNA that was first cut. If we take a step back, you may remember, from the first part of the study aide, that a plasmid is a loop of DNA in a bacteria that is separate from it's 'nuclear' DNA.
This DNA can be passed to different organisms and the traits of the plasmid can incorporate into the nex organism.
For genetic engineering, scientists modify the plasmid and attach more DNA and insert it into the desired organism. For example, a gene from the arctic rod fish is taking and inserted in tomatoes to help them grow in colder environments. This does not make the tomato a non-vegetarian food item. It only allows scientists to insert beneficial traits from one organism into another. Such foods are called genetically modified organisms or GMOs.
So, for example, if the restriction enzyme EcoR1 finds the recognition site on the plasmid and it finds the recognition site on the desired gene of another organism, it will cut. And it will cut the DNA exactly the same, as shown in the table above. Both strands will have sticky ends with 5'AATT3' and 3'TTAA5' hanging off. So, when they come close to each other, H-bonds will form between the complementary sticky ends. What I mean is that the plasmid will have a 3'TTAA5' hanging off and the other strand will have the 5'AATT3' hanging off. These two will come together because of the attractions. Similarly, on the other end, the plasmid will have the 5'AATT3' hanging off and the other strand will have the 3'TTAA5' hanging off. These two will also come together. DNA ligase will form the phosphodiester bonds where they are needed. The bacteria will then move to the desired organism and release the plasmid into it. The plasmid will incorporate itself into the cell's DNA and all the daughter cells will have the desired genes, barring any error.
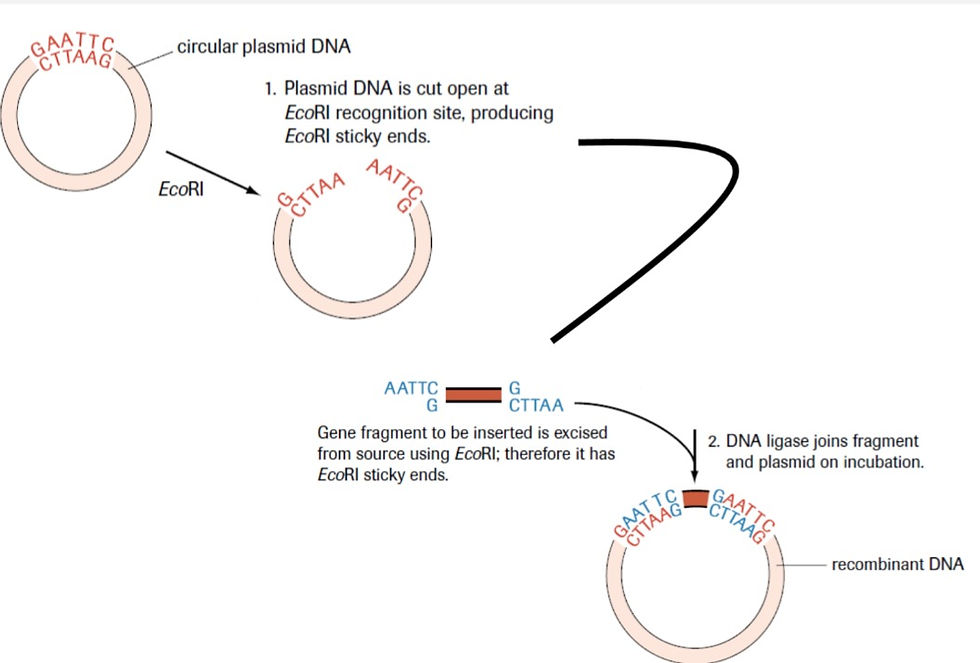
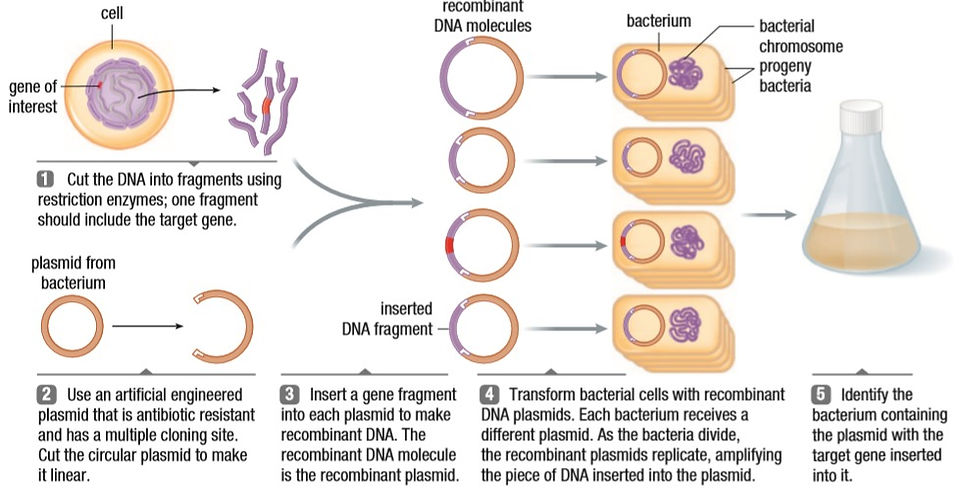
Plasmids are used as vectors in many processes. But not all plasmids pick up on the cut DNA. To increase efficiency, all the involved plasmids are tested for the presence of the required gene. The plasmid with the gene will be replicated multiple times and the new gene will become a part of the plasmid in the daughter cells.
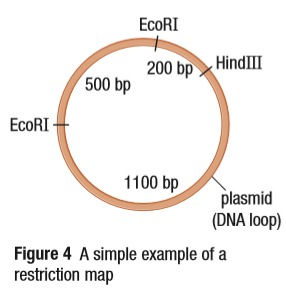
Restriction maps are an interesting concept. They show all the possible recognition sites a plasmid has and the distances between those sites, measured in base pairs. This helps determine which restriction enzyme will be used.

Transformation is probably the most innovative method in this list. The brains it must have taken to think of this process. Transformation is defined as introducing the DNA of a foreign source into a cell. A competent cell, one that can take up foreign DNA, is created in a lab. The bacterial cell is kept in a solution of calcium chloride with the recombinant DNA in an ice bath. The positive calcium ions stabilize the negative phosphate ions in the cell membrane. Once the membrane is physically and chemically stabilized, the solution is quickly heated to 42 degrees Celcius and immediately cooled. This makes the outside of the cell warmer than the inside and water is known to travel for hotter to colder regions. This creates a draft that sweeps the recombinant DNA into the cell. Once the cell is brought back to 37 degrees, the membrane fixes itself. This is harder to do in plant cells because of the cell wall.
There is always the chance that the DNA did not make it into the cell. To test for success, selective plating is done using ampicillin resistance. Ampicillin is an antibiotic and resistance to it allows the bacteria to grow in an environment with ampicillin. The cells are distributed to 4 plates. 2 plates have cells with no resistance; one plate has a medium with no ampicillin and one has ampicillin. The other two plates have cells that are resistant; one plate with a medium without ampicillin and the other with ampicillin. Only the cells with resistance that grow on the ampicillin medium are used for processes. The selective plating shows with cells have the new gene in them. Hopefully, the picture below helps too.
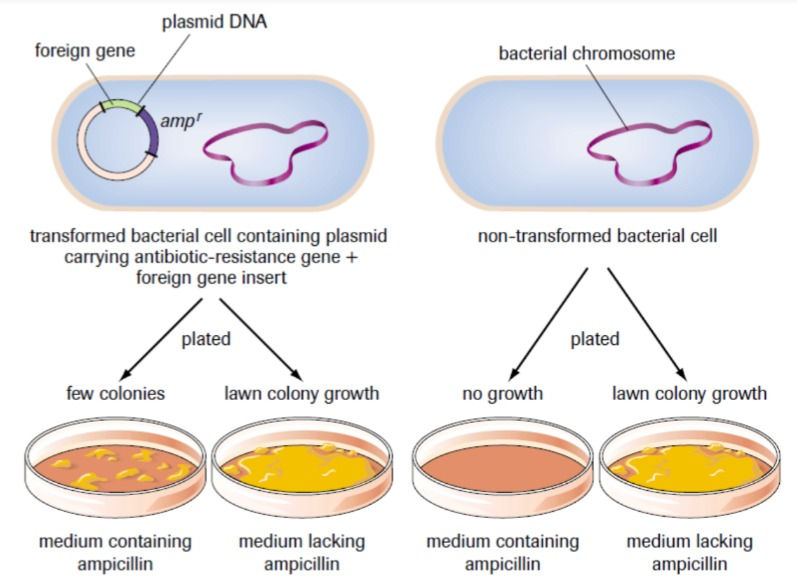
You must remember that the ampicillin resistance is not the gene that is being transferred. It is simply an indicator of whether the cell has a foreign gene or not.
That concludes the manipulation and cloning of DNA.
Next up, DNA Sequencing.
The methods I have been taught about are:
1. Polymerase Chain Reaction
2. Gel Electrophoresis
3. Sanger Dideoxy Sequencing
4. Whole-Genome Shotgun Method
5. Nanopore Sequencing
6. Microarray
In this post, we will talk about the first 3 to avoid the article from being too long to post.
1. Polymerase Chain Reaction
*Fun Fact: we did this whole process at the Science Centre and it was the best field trip of my life*
PCR is a process that makes a large number of copies of a DNA sequence within a few hours using the principles of DNA Replication. Copying the whole genome takes a long time, so only a short sequence is targetted. There are 3 steps: Denaturation, Annealing and Elongation.
1. Denaturation:
In this step, the double strands of DNA are separated. This is done by increasing the temperature to about 94-96 degrees Celcius for 20-40 seconds. This helps break the H-bonds between the complementary bases.
2. Annealing:
For about 20-40 seconds, the temperature is kept between 50-65 degrees. This is a range because the required temperature is dependent on many factors. DNA primers, that are specific to the target, anneal to the single strands of the DNA. The primers are complementary to the strand. They are short and their purpose is to allow the polymerase to attach to the strand. Unlike DNA replication there is no replication fork so one round of replication only requires 2 primers. These primers are specific to make sure that only the target sequence is amplified.
3. Elongation:
The temperature is increased to 72 degrees Celcius. Taq polymerase, a special DNA polymerase found in the archaebacteria Thermus aquaticus, is used for this process because of its heat resistance. The polymerase does its job and complementary strands are made.
What happens is the polymerase only copies until it finds the primer of the other strand. This results in shorter and shorter DNA until they are just the target sequence. Because the first round of replication makes 2 double-stranded DNA molecules, the second step makes 4 molecules, the next makes 8 and it multiplies by 2. This is how so many strands can be made in such a short time.

2. Gel Electrophoresis:
The process is a lot like what the name sounds like. A gel cassette with wells near the tops is used. The DNA solution from the above PCR is dropped into the wells and electricity is passed through the cassette. The DNA is negatively charged so the cathode, or the negatively charged rod, is kept at the top and the anode, or the positively charged rod, at the bottom. The DNA is attracted to the positive charge and moves downwards. The smaller pieces move faster and make it further down the gel than the larger pieces. A molecular marker, also known as a ladder, is kept next to the DNA being tested to see what the lengths of the pieces are. A dye is used to help the DNA move downwards quickly. The DNA is then stained and photographed.
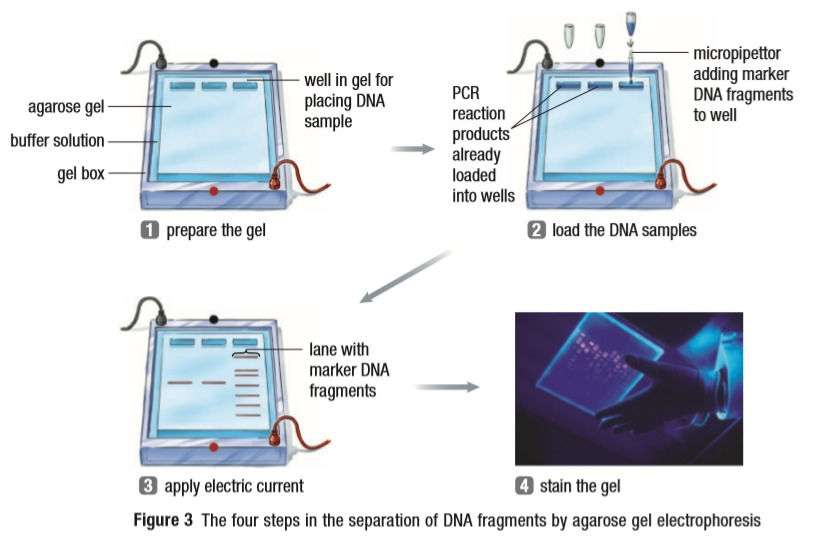
This process is widely used in Forensics, to find a criminal, and Paternity Tests, to confirm paternity.
For Forensics, if a sample of DNA is found at a crime scene, it is compared to the DNA of the suspect(s). If there is a 100% match, the suspect is likely to be guilty. This can't be done with just one sequence because some genes have a higher genotypic frequency, which means they can be very common.

Comment below who you think the criminal is. If you guess right, I'll give you a shoutout in my next post.
Each human has 2 alleles for a gene that they receive from their parents. These alleles can be different, heterozygous, or same, homozygous. When testing for paternity, one allele of the child must belong to the mother and one to the father. If they don't match, the child is likely not their's.

Comment below who you think the father is. If you guess right, I'll give you a shoutout in my next post.
3. Sanger Dideoxy Sequencing:
This process becomes very simple once you have understood it. It uses the principles of DNA replication but the DNA is treated to become single-stranded. A short, radioactive primer of a known sequence is added to the 3' end of the strand. 4 copies of the strand are made and one goes in 1 of 4 test tubes. The test tubes contain a special mix with DNA polymerase, free deoxynucleotide triphosphates (dNTPs) and a very small amount of dideoxy(x)triphosphates (ddNTPs). The x is any of the nitrogenous bases. A regular DNA nucleotide has a deoxyribose sugar where the 2' carbon is missing an -O from the -OH. In a dideoxyribose sugar, the 2' and the 3' carbons are missing the -O. This prevents the 3' carbon from making a phosphodiester bond with the next nucleotide's phosphate and the replicate of that strand stops there.
Each test has dATP, dTTP, dGTP and dCTP in large amounts. Each tube has one of ddATP, ddTTP, ddGTP and ddCTP. What this does, is the dideoxy analogue stops replication in a certain place and scientists can map the DNA strand with that information. The chains made are of different lengths but there are only a finite number of lengths a sequence can have.

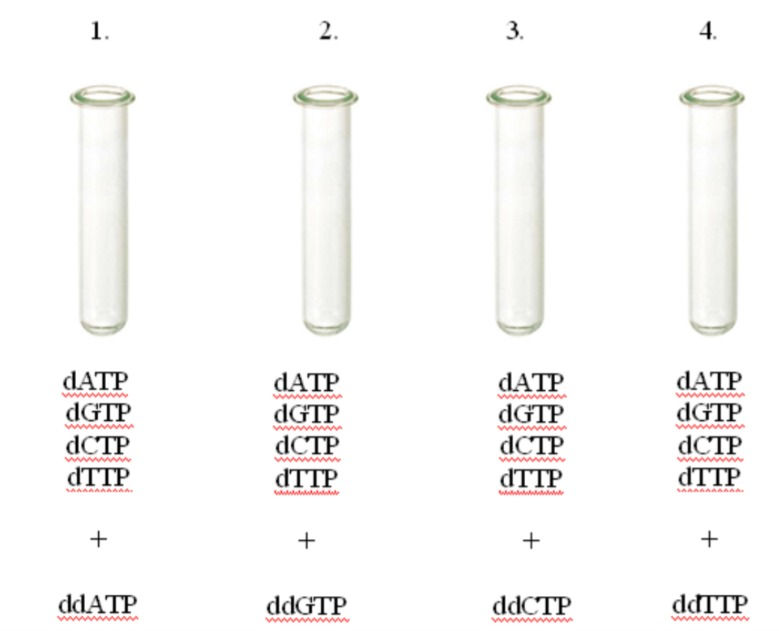

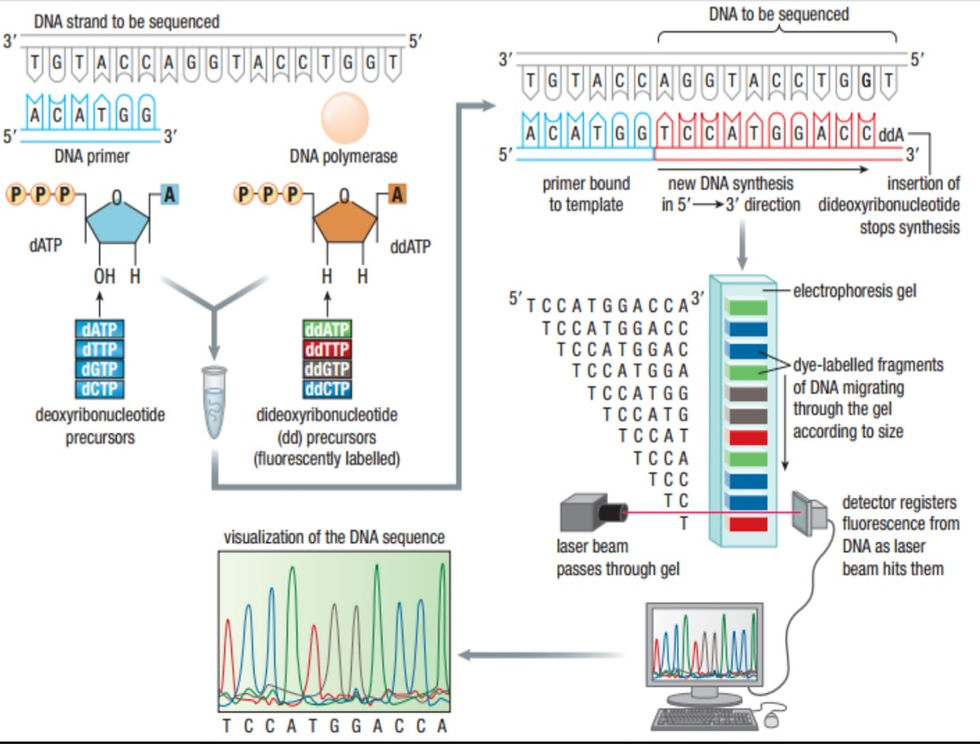
I hope the pictures help you understand what is going on.
This is where I will stop for today. Please let me know if you want me to explain any other topics at or below the Grade 12 Science level. Have a great winter break!-Aarushi
APA Citation:
Fraser, D., LeDrew, B., Vavitsas, A., White-McMahon, M. (2012). Biology 12. Toronto: Nelson Education Ltd.



Comments